
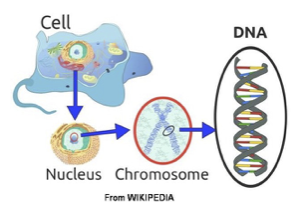
Was ist ein Gen? Wie funktioniert ein Gen?
Jede unserer Zellen enthält 22 + 1 (Geschlechtschromosomen) Paare von Chromosomen. Chromosomen bestehen aus DNS (Desoxyribonukleinsäure) und enthalten daher unsere Erbinformation, unsere Gene. Die DNS ist ein langes Kettenmolekül, das aus vielen Bausteinen zusammengesetzt ist, die man Nukleotide nennt. Jedes Nukleotid hat selbst drei Bestandteile: Phosphorsäure, den Zucker Desoxyribose sowie eine Base. Bei der Base kann es sich um ein Purin, nämlich Adenin (A) oder Guanin (G), oder um ein Pyrimidin, nämlich Thymin (T) oder Cytosin (C) handeln. Die DNS ist normalerweise in Form einer schraubenförmigen Doppelhelix aufgebaut, in der sich zwei oben beschriebene Kettenmoleküle aneinanderlagern. Durch dieses Aneinanderlagern stehen sich in der Mitte der Doppelhelix zwei bestimmte Basen gegenüber, sie sind gepaart (A mit T, G mit C).
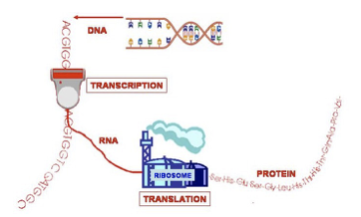
DNS, Transkription, RNS, Ribosom, Translation, Protein
Wie sieht die Struktur eines Gens aus?
Die Struktur eines Gens muss man sich ähnlich wie ein Satz vorstellen. Ein Satz besteht aus Wörtern, die die Information enthalten, und aus Leerstellen, ohne die die zusammengereihten Wörter wenig Sinn ergeben würden. Ein Gen besteht aus Exons, die die Information enthalten (Wörter) und Introns (Leerstellen dazwischen). Bevor der genetische Code abgelesen und in ein Protein umgewandelt wird, werden die Introns herausgeschnitten und die Exons zusammengefügt (diesen Prozess nennt man Splicing).
Interessanterweise, sind die Leerstellen (Introns) zwischen den Wörtern tatsächlich viel länger als die Worte (Exons) selber.
Daher………kann…………………………..ein…………………..Gen………………..so…………….aussehen.
Außerdem sind die Introns keine richtigen Leerstellen. Sie enthalten wichtige Informationen wie das Gen selbst, oder ein anderes in ein Protein umgewandelt wird.
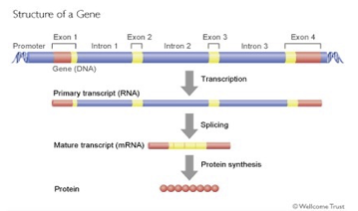
Struktur eines Gens
Das CDKL5-Gen
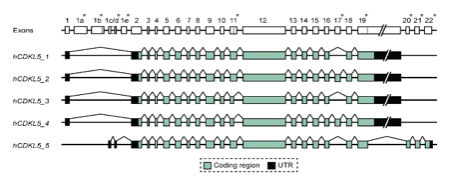
Das CDKL5-Gen liegt auf dem kurzen Arm des X-Chromosoms in Position 22 (Xp22) und besteht aus 24 Exons, inklusive dem Exon 16b, das sich zwischen Exon 16 und 17 befindet. Die 24 Exons enthalten weiterhin die Exons 1, 1a, und 1b, die nicht direkt zu der Struktur des Proteins beizutragen scheinen, da sie nicht translatiert (in Aminosäuren übersetzt) werden. Man zählt daher im CDKL5-Gen 21 Exons (Exon 2-21 – inklusive Exon 16b) die für das CDKL5-Protein kodieren.
Abhängig von Translation der letzten Exons (Exon 17-21), existieren im menschlichen Körper 5 verschiedenen Formen des CDKL5-Proteins. Die prädominante Form im Gehirn ist die Isoform hCDKL5_1, ein 9.7 kb großes Transcript (Exon 1-18).
Und damit zu den Mutationen
Wie vorhin erwähnt enthalten unsere Gene eine Art Code, um das richtige Protein herzustellen. Jeweils drei Basenpaare unserer DNS kodieren für eine bestimmte Aminosäure, einen bestimmten Bestandteil des Proteins. Die richtige Anordnung der Basenpaare ist daher äußerst wichtig, damit die richtige Aminosäure übersetzt und im Protein aneinander gereiht wird. Eine Mutation (ein Fehler in der DNS) verändert die Anordnung der Basenpaare und dadurch kommt es zu einer falschen Abfolge der Aminosäuren, die dazu führt, dass das Protein nicht richtig hergestellt wird.
Studien bei CDKL5-Patienten haben gezeigt, dass es verschiedene Arten von Mutationen gibt.
Punktmutationen: Als Punktmutation wird eine Mutation bezeichnet, in der nur eine Base verändert ist.
Wird bei einer Punktmutation eine Base der DNS gegen eine andere ausgetauscht, so nennt man dies eine Substitution. Das Leseraster wird zwar erhalten, doch der Austausch einer Base, bewirkt den Einbau einer abweichenden, falschen Aminosäure, der zu einem veränderten Protein führt, welches seine Aufgaben nicht mehr vollständig ausführen kann.
Beispiel: „Zahle meine Maut“ wird zu „Zahle meine Maus“
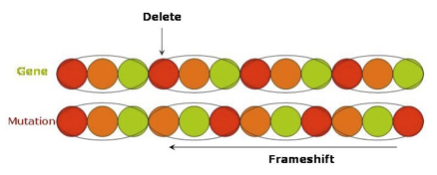
Schlimmer verlaufen kann eine Punktmutation, wenn eine Base komplett wegfällt oder eine neue hinzukommt. Hierbei verliert der DNS-Strang hinter der Mutation seinen ursprünglichen Sinn, da er nach links oder rechts verschoben wird. Man spricht in diesem Fall von sogenannten Frameshift-Mutationen (Leserasterverschiebung). Dies hat zur Folge, dass das translatierte Protein später eine völlig andere Struktur aufweist. Seine ursprüngliche Funktion geht dabei meist verloren.
Deletion: Bei einer Deletion handelt es sich um den Verlust einer Base. Die nachfolgenden Basen rücken gegen die Leserichtung auf, was das Leseraster des nachfolgenden Codons in ebendiese Richtung verschiebt.
Beispiel: „Ich mag nur ein Eis“ >> Deletion der "Base" m >> wird zu „Ich agn ure inE is“
Insertion: Bei einer Insertion handelt es sich um den Zugewinn einer Base. Die nachfolgenden Basen rücken in Leserichtung auf, was das Leseraster des nachfolgenden Codons in ebendiese Richtung verschiebt.
Beispiel: „Ich mag nur ein Eis“ >> Hinter dem Wort „Ich“ wird der Buchstaben X hinzugefügt >> wird zu „Ich Xma gnu rei nEi s“.
Wird durch diese Art von Mutationen eine Aminosäure mit einer anderen ausgetauscht, spricht man von einer sogenannten Missense-Mutation (sinnveränderte Mutation).
Stop-Codon
Ein Stop-Codon ist ein Basentriplette, das sich für gewöhnlich am Ende der Basensequenz befindet und das bei der Proteinsynthese anzeigt, dass das Ende eines Proteins erreicht wurde. Eine Leserasterverschiebung (durch eine Deletion oder Insertion) kann dazu führen dass ein frühzeitiges Stop-Codon "eingebaut" wird. Dies kann überall nach der Mutation passieren und führt zu einer frühzeitigen Beendigung der Proteinsynthese und somit zur Produktion eines verkürzten und nicht funktionierenden Proteins. Man spricht hierbei von einer sogenannten Nonsense-Mutation.
Die oben beschriebenen Deletionen oder Insertionen können natürlich auch mehr als nur eine Base beeinflussen.
Wie werden Mutationen in Berichten beschrieben...
In medizinischen Berichten werden Mutationen generell mit Hilfe von zwei verschiedenen Formaten beschrieben. Das eine Format bezieht sich auf die Basenveränderung (Gen-Mutation), während sich die andere Form sich auf die resultierende Aminosäureveränderung bezieht.
Beispiele:
c.175C>T beschreibt eine Substitution der Base Cytosin (C) an der Position 175 (in Exon 5) mit der Base Thymin (T).
c.2047delG beschreibt eine Deletion der Base Guanin (G) an der Position 2047 (in Exon 14).
c.865insA beschreibt eine Insertion der Base Adenin (A) an der Position 865 (in Exon 11)
Beispiele:
p.Ala40Val beschreibt den Austausch der Aminosäure Alanin in der Position 40 der Proteinkette mit der Aminosäure Valin (p.A40V beschreibt dieselbe Mutation). Diese Veränderung entsteht aufgrund folgender Basenveränderung: c119C>T (Substitution der Base Cytosin (C) an der Position 119 (in Exon 4) mit der Base Thymin (T).
p.R59X. beschreibt den Austausch der Aminosäure Argenin in Position 59 mit einem Stop-Codon (X). Diese Veränderung entsteht aufgrund folgender Basenveränderung: c.175C>T beschreibt eine Substitution der Base Cytosin (C) an der Position 175 (in Exon 5) mit der Base Thymin (T) und führt zu einem verkürzten Protein.
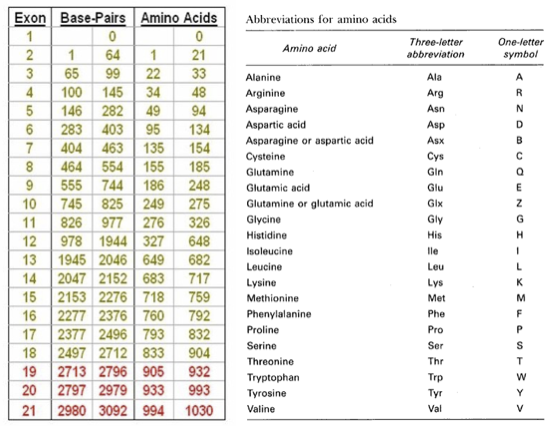
Abkürzungen von Aminosäuren #
Woher kommen Mutationen?
Im Allgemeinen kann es auf 2 Wegen zu einer Mutation kommen. Mutationen können entweder erworben werden oder sie können vererbt werden.
Erworbene Mutationen
Diese Art von Mutationen treten auf, wenn genetisches Material (DNS) auf irgendeine Art geschädigt wird. Solche Fehler geschehen für gewöhnlich während der Zellteilung. Für alle Wachstumsvorgänge im Körper müssen sich Zellen teilen. Dieser Prozess ist sehr komplex, da auch die genetische Information (DNS-Erbinformation) verdoppelt und an die sogenannten Tochterzellen weitergegeben werden muss. Bei solch einem komplexen Prozess kann es leicht zu Fehlern kommen. Man spricht von sogenannten „de novo“-Mutationen, da diese neu auftreten und nicht im Erbmaterial der Eltern des betroffenen Kindes auffindbar sind. Man kann unter anderem zwischen Spontanmutationen und induzierten Mutationen unterscheiden. Spontane Mutationen treten ohne äußere Ursache auf und sind wahrscheinlich auf eingeschlichene Fehler während der Zellteilung zurückzuführen. Induzierte Mutationen sind durch Mutagene (mutationsauslösende Stoffe oder Strahlen) erzeugte Mutationen.
Vererbte Mutationen
Hierbei unterscheidet man zwischen Keimbahnmutationen und somatischen Mutationen. Keimbahnmutationen sind Mutationen, die an die Nachkommen über die Keimbahn vererbt werden, sie betreffen also Eizellen oder Spermien. Diese Mutationen werden von einer Generation zur nächsten übertragen. Somatische Mutationen hingegen betreffen die somatischen Zellen. Sie haben Auswirkungen auf den Organismus, in dem sie stattfinden, werden aber nicht an die Nachkommen vererbt.
Bis heute sind die Ursachen, die zu einer Mutation im CDKL5-Gen führen, unklar. In den meisten Fällen handelt es sich um Spontanmutationen („de novo“-Mutationen), also Fehler, die sich während des komplexen Prozesses der Zellteilung einschleichen. Es gibt vermutlich aber auch vererbte CDKL5-Mutationen. In diesem Fall ist das Gen in den Keimbahnen einer der Elternteile mutiert (Spermien oder Eizellen). Da nur die Keimbahnzellen und nicht die somatischen Zellen betroffen sind, zeigen die Eltern keine Symptome.
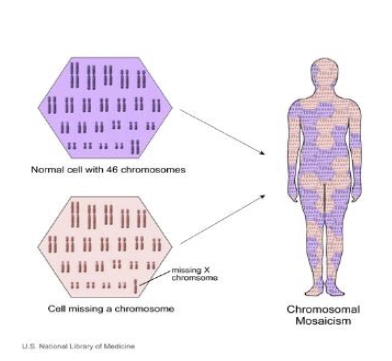
Was ist ein Mosaik?
Wie oben kurz erwähnt, besteht der menschliche Körper aus zwei Typen von Zellen: Keimzellen (Spermien des Mannes und Eizellen der Frau) und somatischen bzw. Körperzellen, die alle anderen Zellen, wie Muskel-, Knochen-, Haut-, Gehirnzellen etc. einschließen. Als Mosaik bezeichnet man in der Genetik ein Individuum, das zwei Zellpopulationen mit unterschiedlicher genetischer Information hat. Die unterschiedlichen Erbinformationen in den Zellen eines Mosaiks entstehen im Laufe der Embryonalentwicklung durch Probleme bei der Zellteilung. Als physiologische Ursache eines Mosaiks kommt auch eine X-Inaktivierung infrage (siehe weiter unten). Eine Zell-population enthält die „normale“ genetische Information, während die andere eine Mutation oder andere genetische Veränderung enthält. Ein Mosaik kann sowohl somatische Zellen als auch Keimzellen betreffen.
Keimzellenmosaik
Beim Keimzellenmosaik ist die genetische Veränderung (Mutation) beschränkt auf die Keimzellen (Spermien des Mannes und Eizellen der Frau), während die anderen Zellen normal sind. In dieser Situation zeigt die betroffene Person, das Elternteil, keinen Hinweis auf eine Erkrankung, da die Körperzellen, die den restlichen Körper bilden, die normale genetische Information enthalten. Dennoch kann die Person die Erkrankung durch ihre veränderten Keimzellen an die Nachkommen weitergeben.
Körperzellenmosaik
Es gibt verschiedene genetische Erkrankungen, denen ein Körperzellenmosaik zugrunde liegt. Die Auswirkungen der Körperzellmosaike werden jedoch nicht immer an die Nachkommen weitervererbt, da die Keimzellen ganz normal sind.
Es gibt allerdings Fälle, in denen es sowohl Körper- als auch Keimzellenmosaike gibt. Bei diesen wird die genetische Mutation an die Nachkommen weitergegeben.
X-Inaktivierung
Als physiologische Ursache eines Mosaiks kommt auch eine X-Inaktivierung infrage. Das CDKL5-Gen befindet sich auf dem X-Chromosom. Männer haben ein X- und ein Y-Chromosom, während Frauen zwei X-Chromosomen haben (eins von jedem Elternteil). Normalerweise ist eines dieser beiden X-Chromosomen ausgeschaltet, ein Prozess, den man "X-Inaktivierung" nennt. Welches der beiden X-Chromosomen ausgeschaltet wird, ist wahllos und zufällig.
Die CDKL5-Mutation befindet sich normalerweise nur auf einem der beiden X-Chromosomen. Per Zufallsprinzip wird eines der beiden X-Chromosomen inaktiviert. Allerdings ist nicht in jeder Zelle das gleiche X-Chromosom ausgeschaltet. So kann es sein, dass in einigen Zellen das X-Chromosom mit der CDKL5-Mutation ausgeschaltet wird, während in anderen Zellen das X-Chromosom mit dem normalen CDKL5-Gen ausgeschaltet wird. Dies beeinflusst theoretisch die Auswirkung und das Krankheitsbild der Mutation. Man vermutet, dass der Krankheits-Phänotyp unter anderem von der Anzahl der Zellen, die das X-Chromosom mit der CDKL5-Mutation nutzen, beeinflusst wird. Diese Theorie muss jedoch noch bewiesen werden.
Gibt es eine Beziehung zwischen Krankheitsverlauf und Mutation?
Bis heute ist relativ unklar, ob es eine Beziehung zwischen Art der Mutation und dem Krankheitsverlauf und den Auswirkungen gibt. Wie oben erwähnt, spielt die X-Inaktivierung wahrscheinlich eine wichtige Rolle. Außerdem scheint die Position der CDKL5-Mutation den Krankheits-Phänotyp zu beeinflussen. Eine wissenschaftliche Arbeit aus Frankreich aus dem Jahre 2011 beschreibt die motorischen Fähigkeiten von 77 CDKL5-Patienten in Zusammenhang mit der Position der Mutation im CDKL5-Gen.
Man kann in dieser Analyse einen Trend erkennen: nur 30% der Patienten mit einer Mutation in Exon 1-11 erlernen das Gehen, während die Zahl bei Patienten mit einer Mutation in Exon 12-21 auf 61% steigt. Es scheint als gäbe es einen Zusammenhang zwischen Mutationen in der Kinasedomäne des Proteins (siehe weiter unten; Exon 2-11) und einem schwergradigeren Krankheitsverlauf. Diese Analyse berücksichtigt jedoch andere Faktoren, wie Mutationsart, Grad der X-Inaktivierung, nicht.
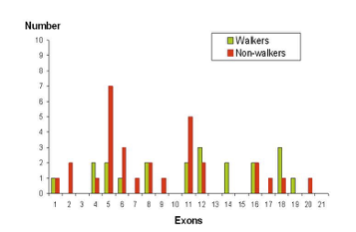
Nummer, Exon, Kasten:Laufen, Nicht-Laufend
Andere klinische Faktoren können auch relevant sein, so wie die Intensität der Therapie jedes einzelnen Patienten oder ob es zusätzliche orthopädische Probleme gibt, wie z.B. Hüft- oder Wirbelsäulenprobleme. Auch können jüngere Kinder eher die Fähigkeit zu gehen entwickeln, während andere, die bereits gehen konnten, ihre Fähigkeit auch wieder verlieren, möglicherweise wegen der schlecht einstellbaren Epilepsie. Darüber hinaus sind die Fallzahlen in dieser Studie relativ klein. Um Antworten auf diese Fragen zu erhalten, wurde kürzlich eine neue Datenbank, die CDKL5 Disorder International Registry Database, entwickelt. Wenn mehr Informationen über Kinder mit dem CDKL5-Gendefekt zur Verfügung stehen, können einige dieser Fragen hoffentlich beantwortet werden.
Text verwendet und übersetzt mit freundlicher Genehmigung von Dr. Martyn Newey, supporting-cdkl5.co.uk, bearbeitet von Claudia Fuchs Phd Neurobiologin an der Universität Bologna, alle Angaben ohne Gewähr.